-
Quantitative Spectra–Structure Relations for Borohydrides
V. D'Anna, L.M. Lawson Daku and H. Hagemann
The Journal of Physical Chemistry C, 119 (38) (2015), p21868-21874


DOI:10.1021/acs.jpcc.5b06045 | unige:76250 | Abstract | Article HTML | Article PDF
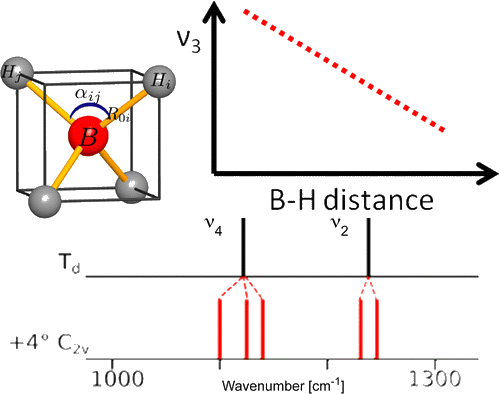
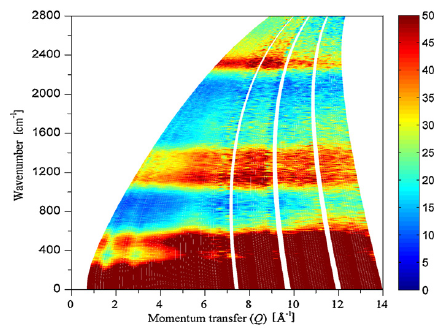
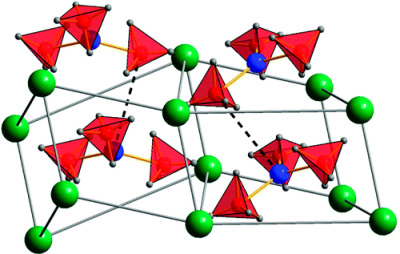
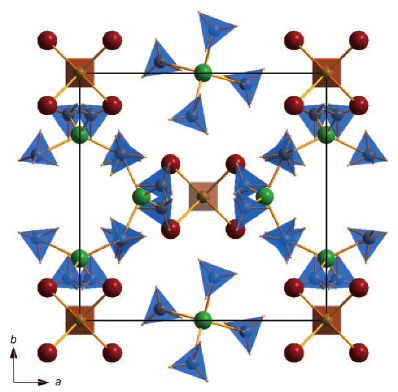
Among the different potential hydrogen storage materials, borohydrides have been largely investigated because of their high gravimetric and volumetric hydrogen content. In the analysis of borohydrides, vibrational spectroscopy plays an important role since it gives information on the local structure of the BH4â ion inside the solid. Here the GF method, developed by Wilson, is used in order to determine the local symmetry of BH4â in solid borohydrides starting from their vibrational spectra. Two different cases of deformations of BH4â are considered. In the first case, the effects of small angular variations on the vibrational spectra of borohydrides will be taken into account; starting from the splitting of the bands corresponding to the deformation modes, the angular deformations will be estimated. In the second one, the BH4â under chemical pressure (in different cubic alkali halides) is considered; in this case, the symmetry of the BH4â remains Td, while the bond lengths change according to the pressure experienced. Different practical examples will be illustrated.